Tutorial 1: Getting Started With ATLAS#
Introduction#
ATLAS is a multi-omics framework that supports trajectory inference on multi-omics data. ATLAS is build upon estimated TI tools for trajectory inference such as Palantir [SKL+19] and CellRank [WLK+24].
In this set of tutorials, you will learn how to:
Construct a multimodal container conatining gene expression and gene activity data
Perform trajectory inference using Palantir as underlying TI strategy
Perform trajecotry inference unsing CellRank as underlying TI strategy
Evaluate results from the trajectory inference
Perform multimodal plots
Data for this tutorial can be found at this link, while cell-type annotations can be found here.
!{sys.executable} -m pip install pysam
Requirement already satisfied: pysam in /Users/lrcq/atlasVenv/lib/python3.12/site-packages (0.24.0)
[notice] A new release of pip is available: 26.0 -> 26.1.1
[notice] To update, run: pip install --upgrade pip
# TO DOWNLOAD DATA IN THE CORRECT LOCATION RUN THIS CELL:
!mkdir -p data/ && cd data \
&& wget https://raw.githubusercontent.com/welch-lab/MultiVelo/main/Examples/cell_annotations.tsv \
&& curl -O https://cf.10xgenomics.com/samples/cell-arc/1.0.0/e18_mouse_brain_fresh_5k/e18_mouse_brain_fresh_5k_filtered_feature_bc_matrix.tar.gz \
&& tar -xvf e18_mouse_brain_fresh_5k_filtered_feature_bc_matrix.tar.gz \
&& curl -O https://cf.10xgenomics.com/samples/cell-arc/1.0.0/e18_mouse_brain_fresh_5k/e18_mouse_brain_fresh_5k_atac_fragments.tsv.gz \
&& curl -O https://cf.10xgenomics.com/samples/cell-arc/1.0.0/e18_mouse_brain_fresh_5k/e18_mouse_brain_fresh_5k_atac_fragments.tsv.gz.tbi
--2026-05-20 13:24:17-- https://raw.githubusercontent.com/welch-lab/MultiVelo/main/Examples/cell_annotations.tsv
Risoluzione di raw.githubusercontent.com (raw.githubusercontent.com)... 185.199.111.133, 185.199.110.133, 185.199.108.133, ...
Connessione a raw.githubusercontent.com (raw.githubusercontent.com)|185.199.111.133|:443... connesso.
Richiesta HTTP inviata, in attesa di risposta... 200 OK
Lunghezza: 135491 (132K) [text/plain]
Salvataggio in: «cell_annotations.tsv.1»
cell_annotations.ts 100%[===================>] 132,32K --.-KB/s in 0,03s
2026-05-20 13:24:17 (4,75 MB/s) - «cell_annotations.tsv.1» salvato [135491/135491]
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 194M 100 194M 0 0 49.2M 0 0:00:03 0:00:03 --:--:-- 49.2M
x filtered_feature_bc_matrix/
x filtered_feature_bc_matrix/features.tsv.gz
x filtered_feature_bc_matrix/barcodes.tsv.gz
x filtered_feature_bc_matrix/matrix.mtx.gz
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 987M 100 987M 0 0 52.2M 0 0:00:18 0:00:18 --:--:-- 53.8M0 0 47.9M 0 0:00:20 0:00:03 0:00:17 47.9M7M 0 0:00:19 0:00:15 0:00:04 51.5M--:--:-- 53.9M
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 709k 100 709k 0 0 4552k 0 --:--:-- --:--:-- --:--:-- 4579k
import os
import atlas
import scanpy as sc
import muon as mu
import numpy as np
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
from muon import MuData
from anndata import AnnData
from scipy.stats import median_abs_deviation
seed = 42
np.random.seed(seed)
data_path = os.path.join(os.getcwd(), "data")
annotation_path = os.path.join(data_path, "cell_annotations.tsv")
ffbcm = os.path.join(data_path, "filtered_feature_bc_matrix")
Multimodal datasets#
ATLAS is built upon the muon.MuData object to operate on paired multimodal informations. The current ATLAS implementation requires at least the following modalities:
rnacontaining scRNA-seq data.One between
atac, containing scATAC-seq data, oractivity, containing gene activity values.
If the activity modality is already present, then ATLAS TI can be directly applied.
If the atac modality is provided, instead, ATLAS performs gene activity computations and requires two additional inputs: the location of a fragment file and the gene coordinates relative to genes whose activity needs to be computed.
In this tutorial, specific anndata.AnnData objects containing scRNA-seq and scATAC-seq modalities are generated and preprocessed before they are merged into a multi-omics object. Genomic coordinates are also retrieved from the gene expression modality.
First, single modalities are read from the 10X genomics input data.
data = sc.read_10x_mtx(ffbcm, var_names="gene_symbols", gex_only=False)
rna = data[:, data.var["feature_types"] == "Gene Expression"].copy()
atac = data[:, ~(data.var["feature_types"] == "Gene Expression")].copy()
ATLAS computed gene activity values from scATAC-seq data via the atlas.pp.preprocessing() function.
Such function requires:
muon.MuDataobject withrnaandatacmodalitiesThe genomic coordinates for the gene of interest
The path to the fragment file.
Genomic coordinates must be provided as an instance of a pandas.DataFrame having at least che following columns:
Chromosomechromosome name, must match the fragments file notationStart: 0-based starting position of the geneEnd: 1-based terminal position for the gene
The DataFrame is here constructed from the features.tsv.gz file provided by 10X Genomics.
Genes are filtered according to the standard chromosomes (1 to 19, X and Y).
valid_chromosome = [f"chr{i}" for i in range(1, 23)] + ["chrX", "chrY", "chrM"]
gene_metadata = pd.read_csv(os.path.join(ffbcm, "features.tsv.gz"), sep="\t", header=None)
gene_metadata.columns = ["id", "symbol", "type", "Chromosome", "Start", "End"]
gene_metadata = gene_metadata[gene_metadata["type"] == "Gene Expression"]
rna.var = (
pd.merge(rna.var, gene_metadata, left_on="gene_ids", right_on="id", how="left")
.drop(["id", "type"], axis=1)
.set_index("symbol")
)
rna.var_names_make_unique()
features = rna[:, rna.var["Chromosome"].isin(valid_chromosome)].var[["Chromosome", "Start", "End"]]
features.head(10)
| Chromosome | Start | End | |
|---|---|---|---|
| symbol | |||
| Xkr4 | chr1 | 3671497 | 3671498 |
| Gm1992 | chr1 | 3466586 | 3466587 |
| Gm19938 | chr1 | 3658903 | 3658904 |
| Gm37381 | chr1 | 3985983 | 3986215 |
| Rp1 | chr1 | 4360313 | 4409241 |
| Sox17 | chr1 | 4496362 | 4497354 |
| Gm37587 | chr1 | 4497473 | 4497474 |
| Gm37323 | chr1 | 4586251 | 4586252 |
| Mrpl15 | chr1 | 4785709 | 4785739 |
| Lypla1 | chr1 | 4807822 | 4807911 |
The path to the fragment file can be provided as input to the atlas.pp.preprocessing() function.
In this tutorial, however, the muon.atac.tl.locate_fragments() function is used to reference the fragment file, so that quality control metrics for the scATAC-seq modality can be computed.
The fragment file describes accessible genomic regions. It is a BGZF-compressed file containing fragment coordinates, with the following minimum required columns:
1st column: fragment chromosome
2nd column: fragment start position
3rd column: fragment end position
4th column: cell barcode
In this tutorial, the fragment file also includes a fifth column reporting the sequencing read support, which ATLAS uses to compute gene activity. If this column is not available, you can set the count_reads parameter to False in the atlas.pp.preprocessing() function, and each fragment will be counted once regardless of its read support.
ATLAS also requires the fragment file to be sorted and indexed (e.g., with Tabix).
fragment_file_path = os.path.join(data_path, "e18_mouse_brain_fresh_5k_atac_fragments.tsv.gz")
mu.atac.tl.locate_fragments(atac, fragment_file_path)
fragments = pd.read_csv(fragment_file_path, sep="\t", header=None, index_col=None)
fragments.head(3)
| 0 | 1 | 2 | 3 | 4 | |
|---|---|---|---|---|---|
| 0 | chr1 | 3000076 | 3000232 | GGCCATCAGGCGCTTA-1 | 1 |
| 1 | chr1 | 3000234 | 3000479 | GAACCAAAGGGACTAA-1 | 2 |
| 2 | chr1 | 3000382 | 3000630 | GTCCGTAAGCCTGTTC-1 | 3 |
ATLAS TI is based on a Weighted Nearest Neighbor (WNN) graph [H+21] computed from gene expression and gene activity.
To obtain a representative WNN, the two modalities must first undergo preprocessing. In this tutorial, we will perform the following steps:
Quality filtering on each modality
Construction of a multimodal object
Log-normalization of gene expression data
Identification of the 2,000 most highly variable genes in the expression space
Gene activity computation
Normalization of gene activity
Dimensionality reduction (PCA)
Single-modality K-Nearest-Neighbor graphs
WNN computation
A comprehensive list of best practices for paired single-cell multiomics data (RNA + ATAC) is available here.
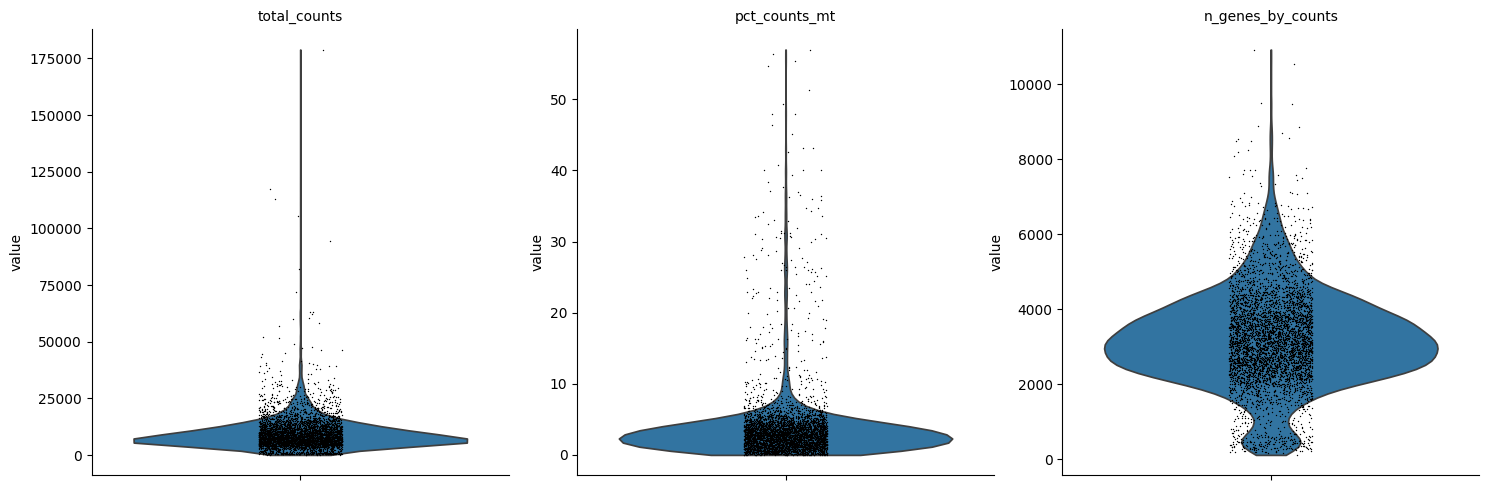
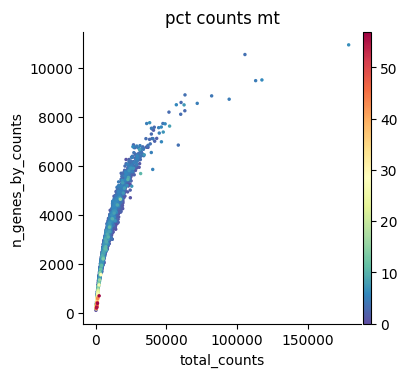
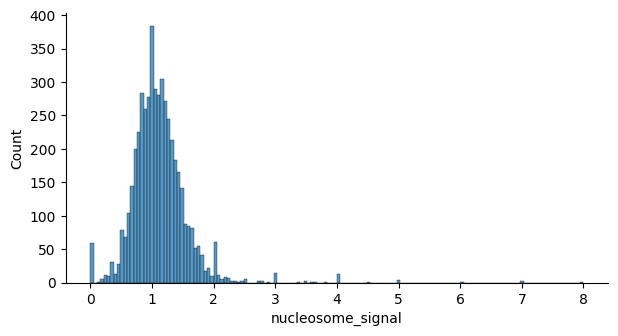
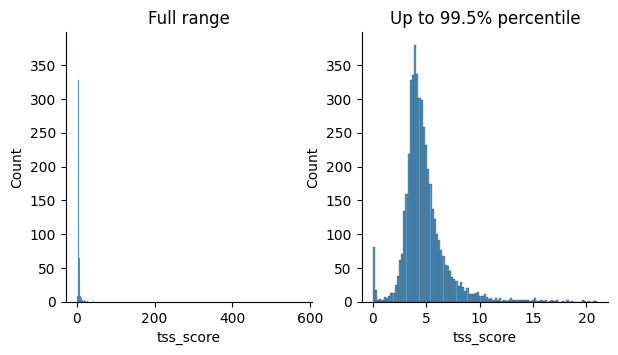
Quality filtering for scRNA-seq data is based on metrics such as the percentage of ribosomal and mitochondrial gene counts and the total number of genes detected per cell. Outliers are identified using the automated approach described here. Quality filtering for scATAC-seq data, instead, relies on modality-specific metrics such as the nucleosome signal and the TSS enrichment score. QC thresholds are chosen based on the distributions shown in the quality control plots below.
We now plot these metrics and identify suitable ranges for filtering. Before filtering, the multimodal object is created. Filtering excludes cells that did not pass quality control in at least one modality. Furthermore, we filter out cells that do not belong to developmental lineages.
rna.var["mt"] = rna.var_names.str.startswith("mt-")
rna.var["ribo"] = rna.var_names.str.startswith(("rps", "rpl"))
sc.pp.calculate_qc_metrics(rna, qc_vars=["mt", "ribo"], inplace=True, log1p=True)
sc.pl.violin(rna, ["total_counts", "pct_counts_mt", "n_genes_by_counts"], multi_panel=True)
sc.pl.scatter(rna, x="total_counts", y="n_genes_by_counts", color="pct_counts_mt")
def _compute_outlier(adata: AnnData, metric: str, nmads: int):
"""Automated outlier detection in scRNA-seq data"""
M = adata.obs[metric]
outlier = (M < np.median(M) - nmads * median_abs_deviation(M)) | (
np.median(M) + nmads * median_abs_deviation(M) < M
)
return outlier
rna.obs["outlier"] = (
_compute_outlier(rna, "log1p_total_counts", 5)
| _compute_outlier(rna, "log1p_n_genes_by_counts", 5)
| (rna.obs["pct_counts_mt"] > 10)
)
sc.pp.calculate_qc_metrics(atac, percent_top=None, log1p=False, inplace=True)
mu.atac.tl.nucleosome_signal(atac, n=1e6)
nuc_threshold = 2
atac.obs["nuc_filter"] = ["NUC_FAIL" if ns > nuc_threshold else "NUC_PASS" for ns in atac.obs["nucleosome_signal"]]
fig, axs = plt.subplots(figsize=(7, 3.5))
sns.histplot(atac.obs, x="nucleosome_signal", ax=axs)
<Axes: xlabel='nucleosome_signal', ylabel='Count'>
tss_enr = mu.atac.tl.tss_enrichment(atac, features=features, random_state=seed)
fig, axs = plt.subplots(1, 2, figsize=(7, 3.5))
p1 = sns.histplot(atac.obs, x="tss_score", ax=axs[0])
p1.set_title("Full range")
p2 = sns.histplot(
atac.obs,
x="tss_score",
binrange=(0, atac.obs["tss_score"].quantile(0.995)),
ax=axs[1],
)
p2.set_title("Up to 99.5% percentile")
Text(0.5, 1.0, 'Up to 99.5% percentile')
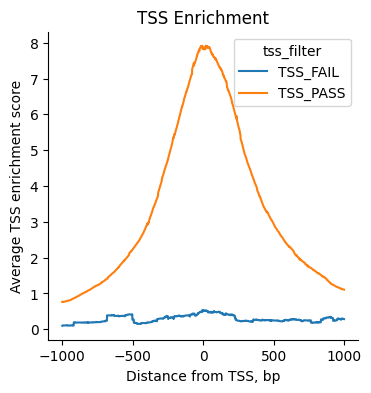
tss_threshold = 1.5
tss_enr.obs["tss_filter"] = ["TSS_FAIL" if score < tss_threshold else "TSS_PASS" for score in atac.obs["tss_score"]]
atac.obs["tss_filter"] = ["TSS_FAIL" if score < tss_threshold else "TSS_PASS" for score in atac.obs["tss_score"]]
fig, ax = plt.subplots()
mu.atac.pl.tss_enrichment(tss_enr, color="tss_filter", ax=ax)
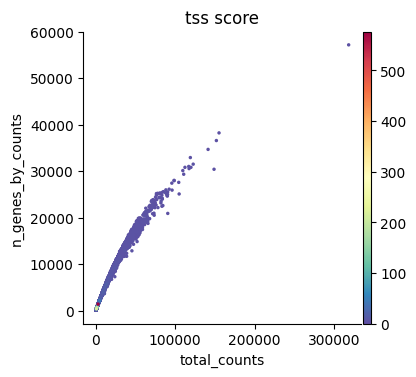
sc.pl.scatter(atac, x="total_counts", y="n_genes_by_counts", color="tss_score")
plot_tss_max = 20
g = sns.jointplot(
data=atac[(atac.obs["tss_score"] < plot_tss_max)].obs,
x="total_counts",
y="tss_score",
color="black",
marker=".",
)
g.plot_joint(sns.kdeplot, fill=True, cmap="Blues", zorder=1, alpha=0.75)
g.plot_joint(sns.kdeplot, color="black", zorder=2, alpha=0.75)
<seaborn.axisgrid.JointGrid at 0x3077de1b0>
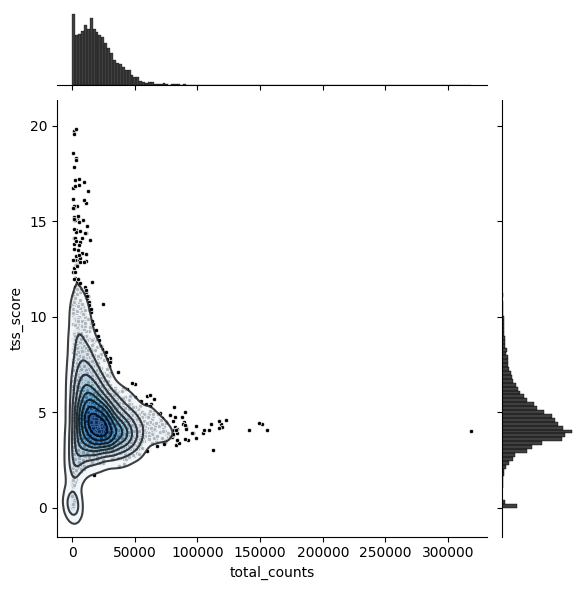
atac.obs["outlier"] = (
(atac.obs["tss_filter"] == "TSS_FAIL") | (atac.obs["tss_score"] > 15) | (atac.obs["nuc_filter"] == "NUC_FAIL")
)
data = MuData({"rna": rna, "atac": atac})
mask = ~(data.obs["rna:outlier"] | data.obs["atac:outlier"])
data = data[mask, :].copy()
annotations = pd.read_csv(annotation_path, sep="\t", header=0, index_col=0)
data.obs = data.obs.join(annotations)
non_developing_clusters = ["Cajal-Retzius", "Interneurons1", "Interneurons2", "Interneurons3", "Microglia", np.nan]
data = data[~data.obs["celltype"].isin(non_developing_clusters)].copy()
sc.pp.normalize_total(data["rna"])
sc.pp.log1p(data["rna"])
sc.pp.highly_variable_genes(data["rna"], n_top_genes=2000)
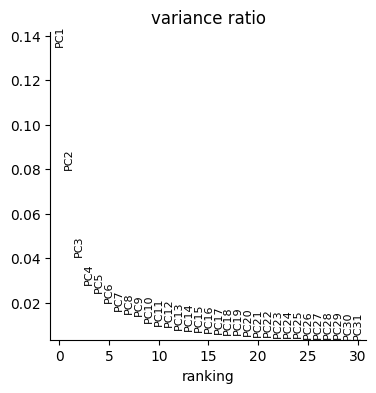
sc.pp.pca(data["rna"], random_state=seed)
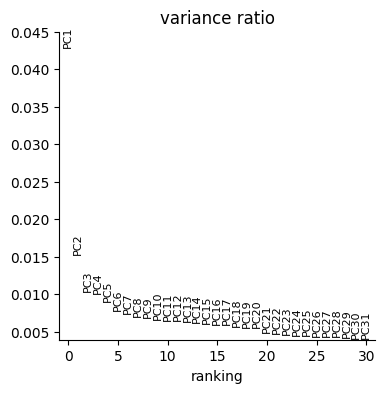
sc.pl.pca_variance_ratio(data["rna"])
ATLAS Preprocessing#
We now obtained all the necessary data and structures to apply the ATLAS preprocessing function. This function automatically perfroms gene activity computation, normalization and dimensionality reduction, as well as the computation of single-modality KNNs and the WNN.
The atlas.pp.preprocessing() requires:
Multiomics data into a
muon.MuDataobjectGenomics coordinates
Path to the fragment file: since the file was previously located in the
atacmodality, we do not explicitly set it.Preprocessing parameters such as the number of PCs and neighbors for single-modality computations. We here chose to search for per-modality 15 neighbors and set the number of PCs to 20 and 10, respectively for gene expression and gene activity modalities.
Parameters for the WNN construction: here we set the number of neighbors to the per-modality arithmentic mean by declaring
wnnparamere equal toNone.
As a result, a separate MuData object containing cell metadata and the rna and activity modalities and intermodality structures is obtained.
knn_rna, knn_act, wnn = 15, 15, None
n_pcs_rna, n_pcs_act = 20, 10
mudata = atlas.pp.preprocessing(
mudata=data,
n_pcs_rna=n_pcs_rna,
n_pcs_act=n_pcs_act,
knn_rna=knn_rna,
knn_act=knn_act,
n_neighbors=wnn,
features=features,
)
mudata
MuData object with n_obs × n_vars = 3486 × 64480
obs: 'atac:n_genes_by_counts', 'atac:total_counts', 'atac:nucleosome_signal', 'atac:nuc_filter', 'atac:tss_score', 'atac:tss_filter', 'atac:outlier', 'celltype'
uns: 'wnn', 'umap'
obsm: 'atac', 'X_umap'
obsp: 'wnn_distances', 'wnn_connectivities'
2 modalities
rna: 3486 x 32285
obs: 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo', 'outlier'
var: 'gene_ids', 'feature_types', 'Chromosome', 'Start', 'End', 'mt', 'ribo', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg', 'pca', 'neighbors'
obsm: 'X_pca'
varm: 'PCs'
obsp: 'distances', 'connectivities'
activity: 3486 x 32195
obs: 'n_genes_by_counts', 'total_counts', 'nucleosome_signal', 'nuc_filter', 'tss_score', 'tss_filter', 'outlier'
var: 'Chromosome', 'Start', 'End'
uns: 'pca', 'neighbors'
obsm: 'X_pca'
varm: 'PCs'
obsp: 'distances', 'connectivities'sc.pl.pca_variance_ratio(mudata["activity"])
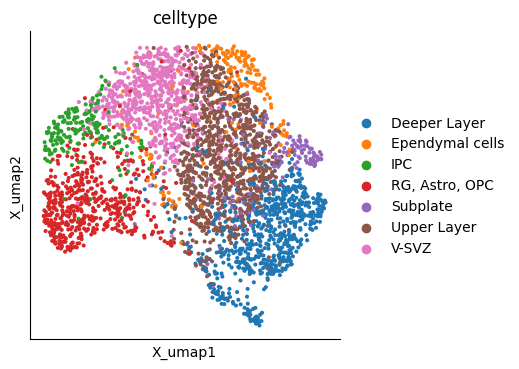
mu.pl.embedding(mudata, basis="X_umap", color=["celltype"])
mudata.write(os.path.join(data_path, "data.h5mu"))